BICON-CEDiT
Extended Hückel Band Structure and
Electronic Dipole induced Transitions Calculations
by Martin Brändle, Ruedi Rytz, and Gion Calzaferri
Department of Chemistry and Biochemistry
University of Bern
Freiestrasse 3, 3012 Bern, Switzerland
Copyright by the Authors
 What is BICON-CEDiT?
What is BICON-CEDiT?
What does the distribution contain?
How do I install the package?
What requirements must my PC fulfil?
How do I run the programs?
Tips and tricks
What is BICON-CEDiT?
BICON-CEDiT is a set of FORTRAN programs that allow extended-Hückel tight-binding calculations on 1D-3D
solid-state compounds, visualisation of results like band structures, density of states (DOS) and crystal
orbital overlap population curves (COOP), and the calculation of oscillator strengths[1],[4] in the crystal.
BICON-CEDiT is based on the EHMACC and EHPC programs of the QCPE #571 package by M.-H. Whangbo et
al.[9]
New features include a two-body repulsive energy term[7],
[8], a distance-dependent weighted Wolfsberg-Helmholz formula
[7],[8], symmetry analysis of the crystal orbitals
[6], use of fractional coordinates, automated geometry variation, and
the calculation of oscillator strengths in the crystal[1],
[4].
BICON-CEDiT consists of five parts, EHMACC, EHPC,
BANDPLOT, DOSPLOT, and CEDiT,
and of some miscellaneous tools.
- EHMACC
- solves the extended-Hückel secular equation for solid state compounds and molecules.
Slater-type s, p, d, and f orbitals, different Wolfsberg-Helmholz approximations,
a two-body repulsive energy term, an automated geometry variation, a fragment molecular orbital FMO
option, and a symmetry analysis of the crystal orbitals are included. Input is easily done by
creating an inputfile containing legible commands, keywords and parameters with the users favourite
editor.
- EHPC
- calculates density of states DOS and DOS-weighted properties like the total energy, the Fermi
level, and the COOP of an infinite solid. The input concept is the same as with EHMACC.
- BANDPLOT
- allows the band interpolation and the graphic representation of bands. The user is guided
through a simple text menu system which enables very fast input and control of the various graphic
parameters. BANDPLOT uses the PGPLOT graphics subroutine library
by T. J. Pearson, California Institute of Technology.
- DOSPLOT
- is used for the graphic representation of density of states weighted properties. The user is
guided through a simple text menu system. DOSPLOT uses the
PGPLOT graphics subroutine library by T. J. Pearson, California Institute of Technology.
- CEDiT
- calculates oscillator strengths of electronic dipole-induced
transitions (EDiTs) in the solid based on Slater-type extended-Hückel wave functions.
- tools
- include k-point sets and programs to generate them, transc for conversion of coordinates,
and dosapp for merging DOS curves.
All programs are written in FORTRAN and run on Windows NT i386 PCs. No machine specific specialities
have been included. The programs should be easily portable to other operating systems. Versions have been
tested on AIX, Linux, VMS, MS-DOS, and MS-Windows. We provide, however, no support for porting the programs.
What does the distribution contain?
 PROGRAMS (exe directory ("working"
directory))
PROGRAMS (exe directory ("working"
directory))
- The 32-bit binaries of EHMACC, EHPC, BANDPLOT, DOSPLOT, and CEDiT compiled by Microsoft FORTRAN
PowerStation 4.0.
- The corresponding batch files *.bat for startup and parameter passing.
- A file grfont.dat containing the Hershey fonts for BANDPLOT and DOSPLOT.
EXAMPLES (examples directory)
- Some ready to use commented example files are
- for band structure calculations
- c_gitter.ehi (square carbon lattice)
- polyb136.ehi (alternating all-trans-polyacetylene)
- polyb143.ehi (non-alternating all-trans-polyacetylene)
- co_100ba.ehi ((100) monolayer of carbon monoxide)
- ni_100ba.ehi ((100) surface of Ni slab consisting of 4 layers)
- mos2band.ehi (slab of molybdenum sulphide[3])
- sodal_ba.ehi (silica sodalite[5])
- diamond.ehi (diamond (3s orbitals added)[2])
- for properties calculations
- c_gdos.ehi, c_gdos.epi (square carbon lattice)
- polyd136.ehi, polyd136.epi (alternating all-trans-polyacetylene)
- polyd143.ehi, polyd143.epi (non-alternating all-trans-polyacetylene)
- co_100ds.ehi, co_100ds.epi ((100) monolayer of carbon monoxide)
- ni_100ds.ehi, ni_100ds.epi ((100) surface of Ni slab consisting of 4 layers)
- co_ni_18.ehi, co_ni_18.epi (CO on (100) surface of Ni, d(Ni-C) = 1.8 Å)
- co_ni_30.ehi, co_ni_30.epi (CO on (100) surface of Ni, d(Ni-C) = 3.0 Å)
- co_ni_45.ehi, co_ni_45.epi (CO on (100) surface of Ni, d(Ni-C) = 4.5 Å)
- mos2prop.ehi, mos2prop.epi (slab of molybdenum sulphide[3]
)
- sodal_ds.ehi, sodal_ds.epi (silica sodalite[5])
- for structure variations
- polyvary.ehi (all-trans-polyacetylene)
- for CEDiT calculations
- polye136.ehi, polye136.edi, polye136.gen (alternating all-trans-polyacetylene)
- mos2osc.ehi, mos2osc.edi, mos2osc.gen (molybdenum sulphide[3])
- Some output files you may use for comparison are
- polyb136.out (compare with your polyb136.eho file)
- polyd136.out (compare with your polyd136.epo file)
- polye136.out (compare with your polye136.edo file)
K-SETS (k_sets directory)
SOURCES (sources directory)
How do I install the package?
You may wish to copy all distributed files to your hard disk in a directory
called c:\biconcedit. If you do not already have the PKUNZIP utility get
it here
and unpack the four archives by typing
pkunzip -d *.zip
What requirements must my PC fulfil?
BICON-CEDiT runs on PC i386 or higher with Windows NT. As all declared data must load into memory at
startup, a compromise between the desire to treat as large systems as possible and the memory requirement had
to be made. We decided to compile BICON-CEDiT for 100 atoms and 400 AOs. Maximum memory requirement is 29 MB.
We recommend at least 32 MB of RAM and enough page file size. If you have less RAM available it is still
possible to run BICON-CEDiT by allowing the programs to page out larger parts of the memory to the hard disk by
increasing the page file size. Another possibility is to reduce the memory requirement of the programs by
recompiling them with smaller array dimensions (see include file sizes of each program).
How do I run the programs?
The order of use of the programs depends on the job one wants to perform.
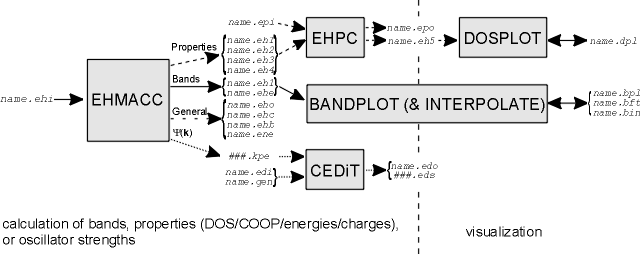
The calculation of band structure diagrams requires the knowledge of the symmetry
properties of the structure to be investigated. First EHMACC has to be called to solve the Schrödinger equation
along several k-points along the high symmetry lines of the Brillouin zone, then BANDPLOT has to be used to
interpolate and plot the bands between the calculated k-points. Properties like total
energy, DOS, or COOP require an EHMACC calculation at many k-points distributed over the irreducible Brillouin
zone. EHPC selects and averages over them, while DOSPLOT is used for displaying the calculated properties.
Oscillator strengths also require their calculation at many k-points distributed over
the irreducible Brillouin zone. EHMACC is used to generate the necessary crystal orbital information, then
CEDiT is called to calculate the oscillator strengths.
Example band structure calculation - Alternating all-trans-polyacetylene
- Open a DOS command prompt window.
- Copy the files polyb136.* in the examples directory to the working directory
exe.
- Change to the exe directory.
- You may examine the input file polyb136.ehi for the band structure calculation. For the
meanings of the keywords cf. chapter 4.1 of the manual.
- Type ehmacc.bat polyb136
- After the calculation has finished, you obtain polyb136.eh1, polyb136.ehc,
polyb136.ehe, polyb136.eho. Examine the output file polyb136.eho.
Compare it with the file polyb136.out to check the correctness of the results.
- An interpolated band structure is provided by the files polyb136.bft and polyb136.bin.
- In the DOS command prompt, type bandplot.bat polyb136
- A new window bandplot is created. Maximize the window. Choose menu Window, entry
Tile. The screen should look like this:
Startup screen of BANDPLOT (Windows NT version).
- At left you see a graphics window, at right a terminal window. Select option (1) in the terminal window
by entering a 1.
- Use the default for the number of symmetry lines. Numbers for energies and symmetry line widths require
a decimal point. Provide an energy range of -35. to 65. eV. The band structure will be drawn, and you
enter a graph modification menu.
- Select option (12). You should obtain a plot like this:
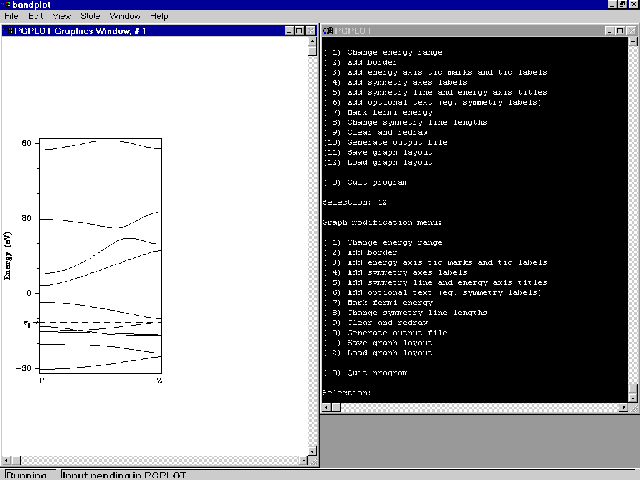
Bandstructure of alternating all-trans-polyacetylene generated by BANDPLOT
- For further explanation of the program have a look at the manual.
Example DOS calculation - Alternating all-trans-polyacetylene
The DOS of all-trans-polyacetylene, its AO projections and the COOP for the C-C and the C-H bonds are
calculated over a mesh of 101 equally dispersed k-points of the linear Brillouin zone.
- Open a DOS command prompt window.
- Copy the files polyd136.* in the examples directory to the working directory
exe.
- Change to the exe directory.
- You may examine the input file polyd136.ehi for the band structure calculation. For the
meanings of the keywords refer to chapter 4.1 of the manual.
- Type ehmacc.bat polyd136
- After the calculation has finished, you obtain polyd136.eh1, polyd136.eh2,
polyd136.eh3, polyd136.eho. Examine the output file polyd136.eho.
- For the integration and smoothing of the DOS and the COOP, EHPC has to be called. First inspect the
input file polyd136.epi. For the meaning of keywords see chapter 4.2 of the manual.
- Type ehpc.bat polyd136
- After the calculation has finished, you obtain the new files polyd136.eh5 and
polyd136.epo. Examine the output file polyd136.epo. Compare it with the file
polyd136.out to check the correctness of the results.
- To visualize the DOS, the DOS projections and the COOP, type dosplot.bat polyd136
- A new window dosplot is created, similar to BANDPLOT. Maximize the window. Choose menu
Window, entry Tile. The screen should look like this:
Startup screen of DOSPLOT (Windows NT version).
- Select option (11). You should obtain a plot like this:
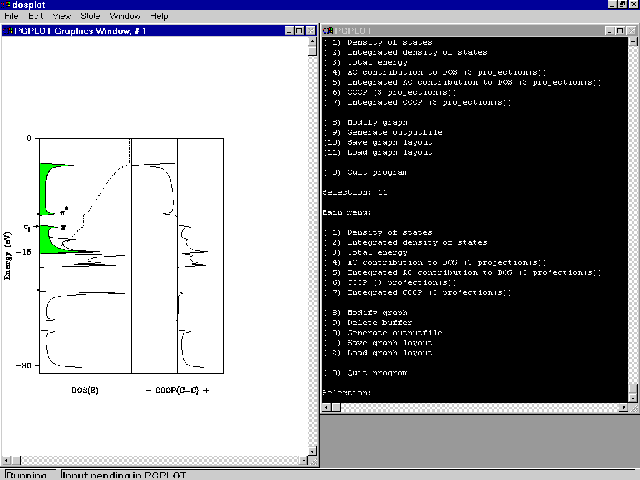
DOS and COOP of all-trans-polyacetylene generated by DOSPLOT.
- For further explanation of the program see chapter 4.3 of the manual.
Example oscillator strength calculation - Alternating all-trans-polyacetylene
The p* ¬ p transitions in the band gap region of a
Peierls distorted all-trans-polyacetylene chain are calculated at 31 equally spaced k-points along
the linear Brillouin zone.
- Open a DOS command prompt window.
- Copy the files polye136.* in the examples directory to the working directory
exe.
- The file polye136.ehi is adapted from polyd136.ehi. For the meanings of the keywords
cf. chapter 4.1 of the manual.
- Type ehmacc.bat polye136
- After successful termination of the calculation you will find the following new files in your working
directory: polye136.eh1, polye136.ehc, polye136.eho, polye136.kp,
and 31 files that are continuously numbered 001.kpe - 031.kpe. They contain the
relevant information for the CEDiT calculation.
- polye136.edi contains the control information for the CEDiT program (cf. Chapter 4.5) of
the manual). Type CEDiT polye136.edo
- A successful calculation will result in the output of the files polye136.edo and
001.eds - 031.eds. Have a look at polye136.edo, where the calculated
oscillator strengths per k-point are given. Compare it with the file polye136.out to
check the correctness of the results.
Tips and tricks
 | BANDPLOT and DOSPLOT: For proper
functioning of the File/Print... and File/Save... menus, set your screen to 256 colors. This
option does not work with 65536 colors. |
References
| [1] | Rytz, R.; Calzaferri G., Oscillator Strengths in Solids:
An Extended Hückel Tight-Binding Approach, J. Phys. Chem. B 1997, 101, 5664. |
| [2] | Calzaferri, G.; Rytz, R., The Band Structure of
Diamond, J. Phys. Chem. 1996, 100, 1112. |
| [3] | Brändle, M.; Calzaferri, G.; Lanz, M., Size
Quantization and Surface States of Molybdenum Sulfide Clusters: A Molecular Orbital Approach, Chem. Phys.
1995, 201, 14. |
| [4] | Calzaferri, G.; Rytz, R., Electronic Transition
Oscillator Strength by the Extended-Hückel Molecular Orbital Method, J. Phys. Chem. 1995,
12141. |
| [5] | Brändle, M.; Calzaferri, G., Silver Clusters in
the Cages of Zeolites: A Quantum Chemical Study, Res. Chem. Intermed. 1994, 20, 783.
|
| [6] | Brändle, M., Moleküle auf inneren und
äusseren Oberflächen, PhD thesis, University of Bern, 1994. |
| [7] | Brändle, M.; Calzaferri, G., Molecular Geometries
by the Extended Hückel Molecular Orbital Method III: Band Structure Calculation, Helv. Chim. Acta
1993, 76, 2350. |
| [8] | Calzaferri, G.; Forss, L.; Kamber, I., Molecular
Geometries by the Extended Hückel Molecular Orbital Method, J. Phys. Chem. 1989, 93,
5366. |
| [9] | Whangbo, M.-H.; Evain, M.; Hughbanks, T.; Kertesz, M.;
Wijeyesekera, S.; Wilker, C.; Zheng, C.; Hoffmann, R., QCPE #571, Extended-Hückel Molecular, Crystal and
Properties Package, Quantum Chemical Program Exchange, 1989. |
